Magnetism
Single Molecule Magnets (SMMs) are low dimensional molecules which can retain the magnetisation upon removal of the magnetic field below a certain temperature known as blocking temperature (TB). The barrier for reorientation of magnetisation is known as Ueff. These magnetically bistable molecules can offer applications like memory storage devices, quantum computing, spin-based electronics, cryogenic refrigeration and etc.
Our group has been working for the past ten years on the theoretical exploration of transition metal based SMMs, lanthanide based SMMs, single-molecule toroics (SMTs), as well as endohedral metallo-fullerene (EMF) based SMMs. With the help of various state-of-the-art quantum chemical techniques such as- Density Functional Theory (DFT), ab initio CASSCF/CASPT2/NEVPT2, Molecular Dynamics and periodic DFT calculations, we aim to study the electronic structure, physical properties, chemical bonding, magnetic exchange, magnetic anisotropy and excited state energetics of transition metal, lanthanide and actinide based systems. We also aim to correlate the structural parameters with the magnetic properties with mapping magneto-structural correlations and modelling new geometries and new structures to predict superior magnetic molecules with enhanced applications in spintronic and data storage devices.
Our major research interests are-
1. Modeling of transition metal based SMM/SIM :
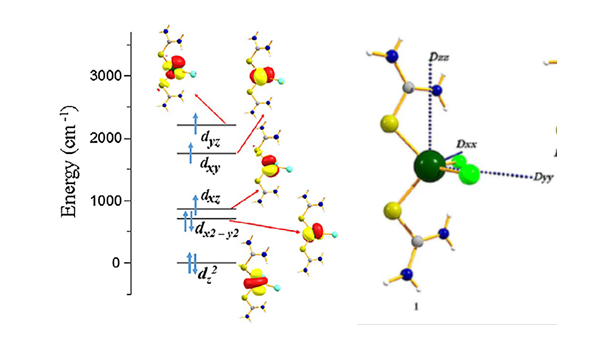
Transition metal based SMMs were first reported in 1993 by Sessoli and co-workers in a {Mn12} cluster. Since then a vast research work have been carried out with various other TM metal mononuclear and polynuclear TM systems in order to achieve large Ueff. Our group has been expertise in this area for a long time specially in mononuclear Ni(II), Fe(II) and Co(II) based single-ion magnets. To estimate the Spin-Hamiltonian parameters of these type of systems, we employ DFT and ab initio methods such as CASSCF/CASPT2/NEVPT2/MRCI techniques. The exchange interactions are obtained from both DFT and multi-reference calculations whereas anisotropy or ZFS parameters we obtain from multi-determinant method only.
2. Lanthanide and Actinde-based SMMs :
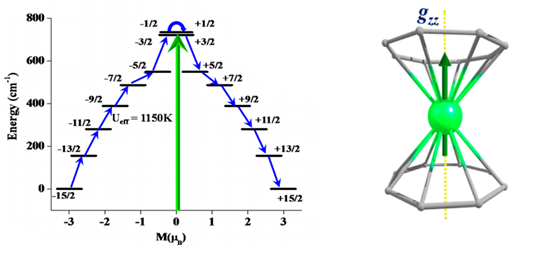
Due to their unquenched spin-orbit coupling lanthanide based SMMs/SIMs are known to possess larger Ueff values. The main challenge in lanthanide based SMMs are controlling the quantum tunneling of magnetisation and other spin-lattice relaxation pathways which lead to under barrier relaxation. From ab initio calculations using RASSCF/RASSI-SO/SINGLE_ANISO modules, we explore and predict theoretical barrier height for reorientation of magnetisation (Ucal) values on existing and as well as model chemical systems. Recently our group is also involved in exploring the spin dynamics of these systems with the help of molecular dynamics. Additionally, we are also explored the effect of external electric field in Ln-based SIMs.
3. 3d-4f systems :
For polymetallic SMMs, the nature of exchange interactions becomes very important as it can lead to a higher ground spin state. Ferromagnetic exchange interactions can also suppress QTM in case of lanthanide-based SMMs and hence to know these interactions becomes highly desirable. Our group has been working in this field for a long time, with hybrid transition metal and lanthanide-based SMMs(3d-4f). These interactions are otherwise difficult to be determined via experimental tools, DFT and ab initio calculations help to understand the electronic structure and nature of magnetic interactions present in the systems.
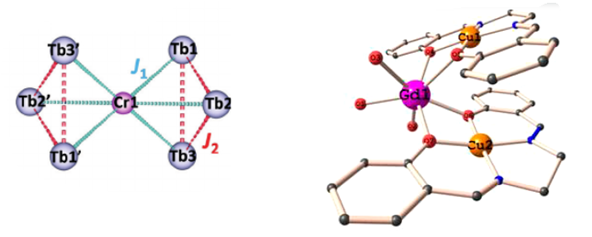
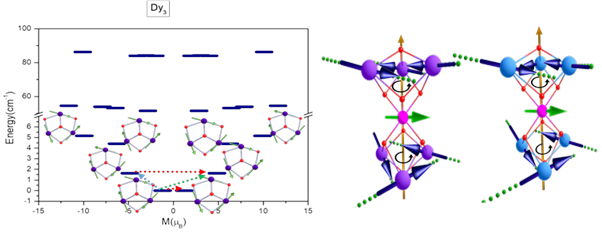
4. Study of Toroidal and Ferro-toroidal unconventional Magnetic systems :
state of the art ab initio calculations has been used to explain the toroidal behaviour of the lanthanide-based triangle, square and six-member ring systems. Our group has been working designing of toroidal and ferro-toroidal systems, their synthesis and characterisation. Single Ln3, Ln4 unit has been proved to show toroidal moment, and the link of transition metal to link to Ln3 triangle has been reported to show the ferro-toroidal behaviour.